Our recent publication “Drivers and sites of variability in the DNA adenine methylomes of 93 Mycobacterium tuberculosis complex clinical isolates”, published in eLife
By analyzing sequencing kinetics of clinical isolates from SMRT-sequencing, we were able to de novo assemble the full genomes and DNA methylomes of clinical isolates spanning seven major MTBC lineages, isolated from several continents. Comparing sequences of de novo assemblies, rather than reference-based variant calling revealed an IS6110 element inserted into and inactivating a methyltransferase that was missed by prior analysis of the same isolate with short reads.
We extend recent methods of DNA methylation heterogeneity analysis and describe “Intercellular mosaic methylation”, a form of prokaryotic DNA methylation heterogeneity distinct from established forms. Though rare overall, intercellular mosaic methylation is nearly ubiquitous in isolates of the globally successful Beijing sublineage suggesting that IMM may confer an adaptive advantage to the Beijing strains.
Integrative analysis revealed widespread promoter methylation in M. tuberculosis, upstream of numerous clinically important genes and operons, and evidence for DNA methylation directly influencing promoter strength. Comparative methylomic analysis distilled 351 hypervariable sites across M. tuberculosis clinical strains, which we propose as potential sites of epigenomic-driven phenotypic variation. This dataset and our findings can provide the basis for future work delineating the roles of DNA adenine methylation in M. tuberculosis physiology and adaptive evolution by the greater research community.
SDSU TB Day 2020
Join us for our SDSU TB day on Friday, February 7th, 2020! The event features tuberculosis experts from around the globe to discuss the burden of TB and its relevance to San Diego’s border health.

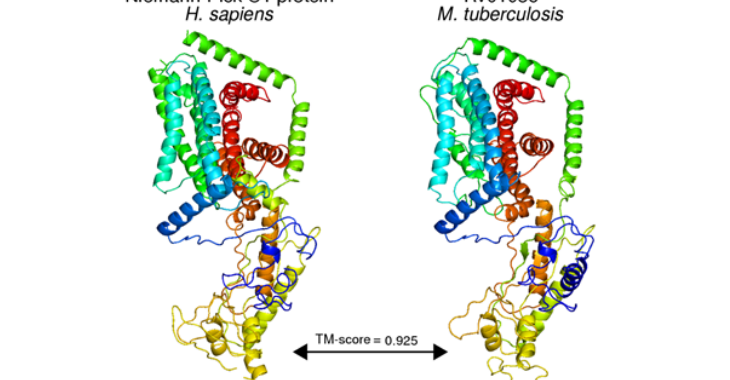
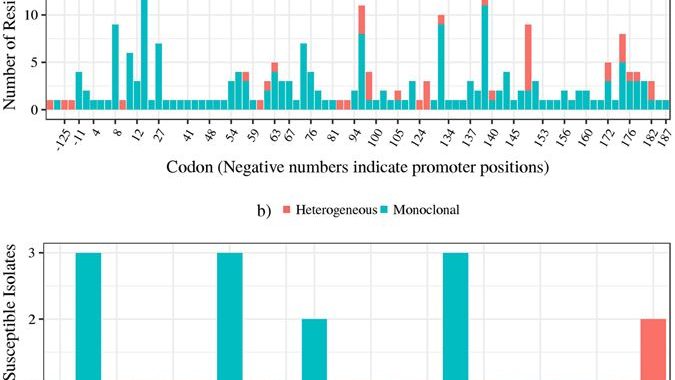
New publication: Pyrazinamide genotypic-phenotypic association in mostly XDR-TB
Pyrazinamide (PZA) is an important first-line drug in all existing and new tuberculosis (TB) treatment regimens. PZA-resistance in M. tuberculosis is increasing, especially among M/XDR cases. Noted issues with PZA Drug Susceptibility Testing (DST) have driven the search for alternative tests. This study provides a comprehensive assessment of PZA molecular diagnostics in M/XDR TB cases. A set of 296, mostly XDR, clinical M. tuberculosis isolates from four countries were subjected to DST for eight drugs, confirmatory Wayne’s assay, and whole-genome sequencing. Three genes implicated in PZA resistance, pncA, rpsA, and panD were investigated. Assuming all non-synonymous mutations cause resistance, we report 90% sensitivity and 65% specificity for a pncA-based molecular test. The addition of rpsA and panD potentially provides 2% increase in sensitivity. Molecular heterogeneity in pncA was associated with resistance and should be evaluated as a diagnostic tool. Mutations near the N-terminus and C-terminus of PZase were associated with East-Asian and Euro-American lineages, respectively. Finally, Euro-American isolates are most likely to have a wild-type PZase and escape molecular detection. Overall, the 8-10% resistance without markers may point to alternative mechanisms of resistance. Confirmatory mutagenesis may improve the disconcertingly low specificity but reduce sensitivity since not all mutations may cause resistance. Link to paper: https://www.nature.com/articles/s41598-017-03452-y